

Dr. Stefano Scoglio, Ph.D.
Siamo ora passati alla fase delle varianti del virus, presentati come la grande novità negativa di questo 2021. Ma in realtà, anche all’interno di chi crede all’esistenza di questo virus, si parla da sempre di varianti, e ci sono numerosi studi che confermano le continue mutazioni del virus. Quando hanno “isolato” (in realtà sequenziato) il virus in Italia, hanno subito detto che era diverso da quello di Wuhan, che come ho dimostrato in altri scritti, era già sin dall’inizio diverso da sé stesso, essendo nato uno e trino (i ricercatori cinesi hanno affermato di avere trovato tre virus diversi in 3 pazienti, ma essendo simili hanno deciso di chiamarli tutte e tre nCov2, poi divenuto SARS-Cov2). Perché allo Spallanzani o al San Raffaele allora non hanno parlato di variante italiana? Mettendone in luce il diverso livello di infettività e mortalità? Perché non hanno parlato di variante USA quando hanno depositato delle sequenze diverse e uniche del virus in concomitanza con l’esplosione di positivi e morti nelle RSA degli USA, che ha reso gli Stati Uniti, grazie al contributo degli Stati democratici (New York e California), il nuovo centro della pseudo-pandemia? Perché allora c’erano gli asintomatici positivi, e non c’era bisogno di trovare nuovi argomenti per continuare a tenere le popolazioni sufficientemente terrorizzate.
Al GISAID, la banca dati dei virus, a Febbraio 2021 c’erano oltre 450.000 sequenziamenti diversi del virus, ovvero oltre 450.000 varianti. Possibile che solo quella inglese e sudafricana e solo nel 2021, avessero questa natura così maligna da generare nuovo terrore nella popolazione? No, non è possibile, anche perché il virus è un entità inorganica nanomolare, infinitesimale, e come può sopportare quasi mezzo milione di varianti senza che tali varianti si assomiglino o si sovrappongano? E’ tutto completamente ridicolo, e stiamo per vedere quanto sia ridicolo il modo di determinare e rilevare le varianti, inglesi o altro che siano. Talmente ridicolo da potere dar vita ad una nuova categoria del pensiero: il “ridicolo scientifico”.
Come sempre, tutto ha inizio in collaborazione con la OMS, e anche in questo caso con la filiale cinese. Infatti, l’annuncio dell’emergenza della nuova variante è già pieno di equivoci. Il GISAID il 23 Dicembre annuncia che
“Il Regno Unito ha riportato una nuova variante, chiamata VUI 202012/01 (Variante sotto Investigazione, Anno 2020, mese 12, variante 1) (https://www.gisaid.org/references/gisaid-in-the-news/uk-reports-new-variant-termed-vui-20201201/)
Tutto accade attorno al 15-20 Dicembre. In data 18 Dicembre si riunisce il comitato NERVTAG, di cui fa parte anche il famigerato Prof. Ferguson dell’Imperial College, quello che ha fatto le catastrofiche predizioni farlocche sull’impatto della pandemia, che hanno portato ai vari lockdown nei diversi paesi; salvo poi essere stato scoperto a violare il lockdown, che lui stesso promuoveva, per andare a trovare un’amante (Lucianne.com News Forum – Neil ‘Lockdown’ Ferguson Gets Caught<br> With His Pants, And His Credibility, Down). Il NERVTAG, pur non avendo “…dati insufficienti per trarre qualsiasi conclusione…” sulle questioni fondamentali della variante,
“…ha una moderata sicurezza che la variante VUI-202012/01 dimostri un sostanziale aumento della trasmissibilità in rapporto ad altre varianti.” (NEVRTAG Meeting on SARS-Cov-2 variant under investigation VUI-202012/01, 18 December 2020.).
Sempre nello stesso periodo, il 20 Dicembre, viene sottoposto un articolo a Eurosurveilance (che lo pubblica il 7 Gennaio 2021), da parte di ricercatori cinesi e di Hong Kong legati alla OMS. Il fatto che sia stato sottoposto a pubblicazione nello stesso tempo in cui veniva portato alla luce mostra chiaramente uno sforzo coordinato. Ed è interessante vedere come l’ipotesi della nuova variante sia giustificata da questa equipe cinese. Lo studio afferma che ci sono state due varianti nel Regno Unito, una in Galles (501Y Variant 1) e una in Inghilterra (501Y Variant 2). Delle due, la seconda avrebbe dimostrato di essere del 75% più trasmissibile della originale variante 501N, che sarebbe la matrice da cui sono emerse le due varianti. Sarebbe questa elevata trasmissibilità che ha fatto si che
“…questa variante è diventata il ceppo dominante del virus in Inghilterra nel periodo Novembre/Dicembre 2020.” (Leung K. et al., Early transmissibility assessment of the N501Y mutant strains of SARS-Cov2 in the United Kingdom, October to November 2020, in Euro Surveillance, 2021: 26(1), p. 3.)
A parte il fatto che non si capisce perché la comparazione è stata fatta con la variante originaria e non con l’originale SARS-Cov2 (forse perché non c’è?); non si capisce neppure come abbiano fatto a fare questi calcoli. I primi sequenziamenti della variante sono stati fatti a Dicembre, e nella riunione del 18 Dicembre del NERVTAG viene affermato “…che può essere complicato sequenziare la variante VUI-202012/12/01…”. Come hanno fatto i ricercatori cinesi a calcolare la crescita della nuova variante a partire da Ottobre? Udite, udite! Hanno calcolato il numero di sequenziamenti della nuova variante depositati presso il GISAID nel periodo 22 Settembre-1° Dicembre 2020! Cioè, non c’è nessuna relazione con l’effettiva circolazione di una nuova variante nella popolazione, l’unico dato è quello relativo alla scelta dei sequenziamenti fatta dai laboratori inglesi, e quindi al numero delle sequenze geniche depositate, mostrato nel grafico seguente :
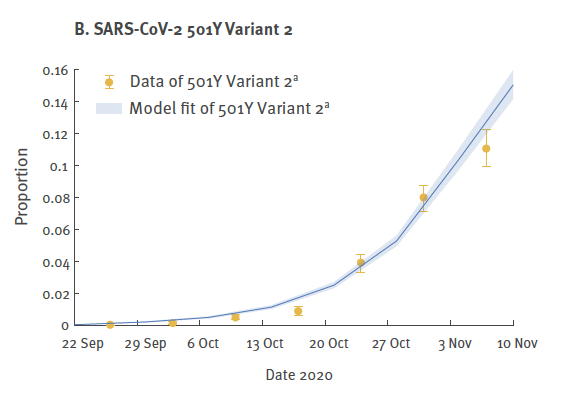
E’ evidente che questo dato, lungi dall’essere un sintomo dell’aumento della diffusione della variante inglese, mostra solo come i pochi ed esclusivi laboratori inglesi che fanno sequenziamenti genici, a partire da Ottobre hanno deciso di puntare sul lancio di una nuova variante! Cosa questa che viene riconosciuta dagli stessi ricercatori cinesi quando scrivono:
“…la nostra analisi comparativa sull’efficienza delle diverse varianti si è basata sui dati di sequenziamento presentati al GISAID, ed è quindi soggetta al pregiudizio (bias) della scelta di sequenze sottoposti al database pubblico.”
Nonostante questo “pregiudizio” che sta alla base dell’analisi che afferma l’emergenza di una nuova variante, i ricercatori non hanno problemi ad affiancarsi alle autorità sanitarie inglesi e al NERVTAG nel chiedere
“…più rapide e stringenti misure di controllo…necessarie a sopprimere la diffusione, che è esattamente quello che ha fatto il governo del Regno Unito il 19 Dicembre, con l’introduzione di un nuovo livello 4 di restrizioni.”
La cosa è persino troppo ridicola per essere presa sul serio, e quindi ci si aspetterebbe che le autorità sanitarie inglesi e il NERVTAG abbiano qualcosa di più sostanzioso da offrire. Nel documento ufficiale di Public Health England (PHE) si dice che il tutto è iniziato l’8 Dicembre, quando si sono verificati i dati del periodo 10-18 Novembre nel Kent, e si sono trovati 117 casi “geneticamente simili”, senza specificare in cosa sarebbe consistita tale similarità genomica. Dei 6130 casi valutati, solo per il 4% si avevano i genomi disponibili, e tra questi si sono selezionati 117 casi simili (circa un ridicolo 1.9% del totale dei casi). Qui emerge già il problema della selezione, quel pregiudizio che abbiamo visto sopra, quell’ossessione dei centri che sequenziano di puntare tutto sulla variante 501Y-2: ci sono 6130 casi, e si selezionano solo i 117 (l’1.9%) con i sequenziamenti simili, in modo da tralasciare tutte le altre possibili varianti che un test del tutto casuale come la PCR per SARS-Cov2 sicuramente genera.
Ma come si passa da questi primi 117 casi a poter affermare che la nuova variante sta diventando dominante nella popolazione di Londra e del Sud-Est dell’Inghilterra? Il documento PHE afferma che il gruppo del Kent faceva parte di un più ampio cluster nazionale di 915 individui con genomi virali disponibili. Stiamo parlando di numeri ancora irrisori. Il passaggio successivo è quello che si riferisce alla raccolta, a livello nazionale, di 35.211 casi con genomi disponibili: di questi, 1.419 avevano un genoma inclusivo della nuova variante, mentre 33,792 avevano genomi privi della nuova variante, che viene definita come variante della proteina Spike del virus, tanto che i casi sono descritti come Spike gene target failure (SGTF), ovvero “fallimento del target genetico Spike”. Quindi, al di là del fatto che si sta sempre parlando non di virus isolato ma di sequenziamenti computerizzati, siamo di nuovo a un livello percentuale ridicolo, solo il 4% dei soggetti il cui genoma è stato analizzato avrebbe avuto la variazione, il 96% non ha quella variazione, probabilmente ne avrebbe avute tante altre, se solo avessero volute cercarle.
PHE fa poi una proiezione molto discutibile. Senza spiegare su quali basi, PHE afferma che questa variante VOC (Variant of Concern – il nuovo nome della variante VUI-202012/12/01) genera un aumento del parametro di trasmissibilità Rt di 0.52, cioè del 52%, concludendo:
“…un’area con un Rt di 0.8 senza la variante avrebbe un Rt di 1.32…se la sola variante VOC fosse presente.”
Qui la manipolazione è massima: partono dai 1419 casi su 35.211, ovvero dal 4%; e lo proiettano sull’ipotesi del 100% dei casi (la sola variante VOC presente), arrivando così all’aumento molto significativo del 52% (da 0.8 a 1.32). Ma la versione realistica, il calcolo corretto, dovrebbe essere diverso: si dovrebbe calcolare solo il 4% dell’aumento dello 0.52, che è uguale a 0.0208; e quindi si passerebbe dallo 0.8 allo 0,802, dall’80% all’80,2%! Cioè il vero impatto della variante, ammesso e non concesso che esista, sul Rt sarebbe inferiore all’ 1%, una percentuale ridicola.
PHE insiste affermando:
“Una differenza del 10% nella frequenza della variante VOC a metà Novembre corrisponde approssimativamente a un aumento settimanale di 50 casi su 100.000 all’inizio di Dicembre.”
Dato che il tasso più realistico di aumento del Rt è, come abbiamo visto, al massimo dell’1%, l’impatto di questa terribile variante sarebbe di 5 casi su 100.000, una percentuale dello 0.005%: che paura!
Ma dove si tocca l’apice del tragicamente ridicolo è il metodo con cui vengono rilevati i casi di variante inglese nella popolazione. Abbiamo visto sopra come il NERVTAG affermi che sia “…complicato sequenziare la variante…”. Infatti, per sequenziare il genoma del presunto virus e trovare le modifiche geniche che caratterizzano la variante, ci vogliono laboratori specializzati e un paio di settimane. Così, quando ho sentito affermare dai vari organismi sanitari italiani che ormai la variante circola ampiamente, ed ha infettato almeno un 1/5 della popolazione, mi sono detto: 1/5 della popolazione corrisponde a circa 12 milioni di persone; possibile che abbiano fatto il sequenziamento a 12 milioni di persone, e in così poco tempo? No, non è possibile, e infatti cercando ho scoperto che gli inglesi prima, e tutti gli altri poi, hanno deciso di usare non l’analisi genomica ma quello che si chiama un “proxy marker”, un parametro sostitutivo che i laboratori possano usare per rilevare la mutazione:
“Tre di questi laboratori usano un saggio con 3 target (N, Orf1ab, S) con il sistema TermoFisher (TaqPath).”
Si tratta di un sistema PCR che analizza 3 geni, il gene N (nucleocapside), il gene S (Spike) e il gene Orf1ab). Si ricorderà che mentre all’inizio i tamponi PCR davano positività solo se si trovavano tutti e tre i geni, da Aprile, in seguito a un comunicato della OMS, è bastato trovare un solo gene (con tutti i problemi di falsi positivi annessi). Ora, le autorità sanitarie inglesi (PHE) dicono che per rilevare la variante VOC (VUI-202012/01) si può usare la negatività al gene S come indicatore sostitutivo della presenza della variante:
“Quindi, usiamo la frequenza della negatività al gene-S tra i positivi alla PCR come indicatore sostitutivo della frequenza della VOC.” (Public Health England, Investigation on novel SARS-Cov2 variant. Variant of Concern 202012/01, Published: December 2020, PHE gateway number: GW-1824, p. 3.)
Veniamo subito al punto: ci stanno dicendo che la negatività al gene S, con parallela positività ad uno degli altri due geni, ovvero quello che fino ad oggi era stata una normale positività, è ora diventata la prova dell’esistenza della variante. Vorrei sottolineare ulteriormente la presa per i fondelli di questa variante: fino a Dicembre 2020, in tutto il mondo, se eri positivo al solo gene N o al solo gene Orf1, eri semplicemente positivo. A partire dalla invenzione della nuova variante inglese, se sei positivo al solo gene N o Orf, sei positivo alla nuova variante inglese.
In altre parole, la variante inglese si fonda su una doppia truffa: primo, perché come è stato sottolineato da più parti, perché ci sia vera positività occorre che tutti e tre i geni siano positivi, che solo così si potrebbe rilevare un virus intero e dunque attivo, altrimenti ne trovi solo dei pezzi che potrebbero benissimo essere residui di un virus passato ed eliminato. Ma ora, invece di correggere questo errore manipolativo, ci hanno aggiunto un altra truffa: se questa volta, invece di essere positivo al solo gene S, sei positivo al solo gene N o Orf, cosa che fino a Dicembre ti rendeva semplicemente (e ingiustamente) positivo, sei dichiarato affetto dalla variante inglese!
Che si tratti di un metodo ulteriormente truffaldino, di fatto lo ammette anche il PHE:
“Questo indicatore sostitutivo ha una finestra di validità limitata, ed è generalmente un sostituto inefficace più indietro si va nel tempo, a causa di altre vecchie varianti che testavano anch’esse negative alla proteina spike.”
Certo, se vai nel passato, i test negativi alla proteina Spike, ovvero al gene S, erano già stati catalogati come semplicemente positivi, e quindi si sarebbe dovuti andare riclassificarli tutti come variante inglese, cosa assurda dato che la variante VOC ancora non si era sviluppata.
A questo livello di ridicolo, impunemente accettato da tutti i media e i politici senza batter ciglio, si aggiunge un ulteriore fattore: PHE ammette qui che esistono “vecchie varianti” che rispondono positivamente a uno degli altri due geni e negativamente al gene S. Qui PHE parla al passato, ma altrove nel documento parla di varianti attuali:
“Molto meno si sa di altre varianti della proteina spike presenti nel gruppo, con l’eccezione della variante D614G che è stata ben caratterizzata ed è già prevalente nel Regno Unito. Il loro significato al momento non può essere giudicato.” (ibid., p.7)
Quindi, nel passato l’indicatore sostitutivo dell’assenza del gene S non vale, perché ci sono altre varianti che potrebbero confondere; ma nel presente e nel prossimo futuro, pur essendoci altre numerosi varianti, sempre della proteina spike/gene S, di cui non sappiamo nulla, questo non rappresenta un problema. Come sempre, nel contesto del ridicolo scientifico, si può sempre avere la botte piena e la moglie ubriaca.
Il PHE approfondisce poi la parte genomica, e mostra la tabella delle varianti VOC:
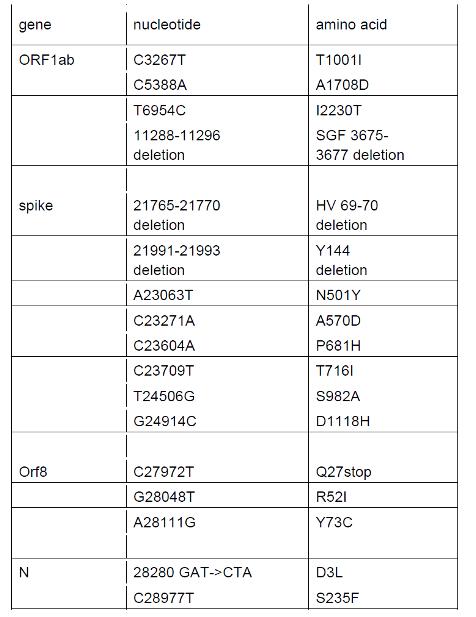
Questa è la Tabella 1, dove vengono definite le mutazioni geniche che caratterizzano la variante, che in base a questa definizione vien ora chiamata B.1.1.7 (dato importante, come stiamo per vedere). La prima cosa da notare è che, mentre PHE & C. puntano tutto sulle varianti del gene S, in particolare la variante N501Y e la deletion (cancellatura) degli aminoacidi HV 69-70 (Δ69-70), da questa tabella emergono anche 5 mutazioni del gene Orf1ab e 2 mutazioni del gene N. Questo pone un altro problema: dato che per essere positivi alla nuova variante bisogna essere negativi al gene S ma positivi agli altri due geni come tradizionalmente definiti dalla PCR storica, cioè coi parametri definiti all’inizio della pseudo-pandemia, sembra chiaro che il fatto che anche questi due geni sono mutati rende l’attuale PCR del tutto casuale ed invalida, dato che, non potendo trovare la positività dei mutati geni N e Orf1ab della variante, finirà per trovare la positivirtà di geni du qualche altra variante, in modo del tutto casuale. Anche questo conferma che la rilevazione della nuova variante non ha alcun valore.
Abbiamo visto come PHE ammetta che esistono altre varianti con ulteriori mutazioni, e di cui non abbiamo sufficienti dati. Ma PHE ammette un dato ulteriore particolarmente significativo:
“Anche alcune varianti diverse dal VOC hanno la Δ69-70…”
Quindi il test che non rileva il gene S a causa di tale deletion (cancellazione) potrebbe in realtà trovare anche altre varianti con tale mutazione e del tutto innocue. PHE cerca di superare questa difficoltà mostrando come la quasi totalità dei test positivi ma con la negatività del gene S si riferiscano specificamente alla variante VOC/B.1.1.7:
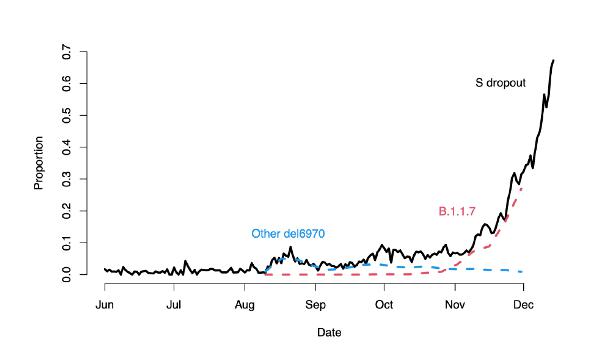
La linea blu rappresenta la frequenza delle altre varianti; quella rossa la frequenza della variante VOC/B.1.17. Come si vede, ed è questa la ragione della pubblicazione di questo grafico da parte di PHE, fino agli inizi di Novembre tutte le varianti avevano la stessa frequenza, anzi le altre varianti predominavano. Poi, a Novembre la variante B.1.1.7 prende il sopravvento, e PHE dichiara che arriva a costituire addirittura il 96% delle varianti!
Ma anche qui c’è il trucco, e come dice il proverbio, il diavolo fa le pentole ma non i coperchi. Il 96% è infatti calcolato sula somma totale delle frequenze blue e rosse. Ma se si guarda bene il grafico, si vede che comunque la linea rossa VOC/B.1.1.7 (variante inglese) arriva fino alla proporzione 0.3 (30%), mentre il totale delle negatività al gene S (S Dropout) arriva fino allo 0.7 (70%). Questo significa che la maggioranza di quelli che testano positivi ad uno degli due geni ma negativi al gene S non hanno la variante inglese: solo il 30% avrebbe questa presunta, e mai dimostrata, variante. Tuttavia, tutti quelli che saranno positivi a un gene, ma non al gene S, anche il 70% di casi che non hanno la variante VOC/B.1.17, verranno dichiarati essere affetti dalla variante inglese, anche se di fatto non lo sono. Questo, di nuovo, significa una notevole maggioranza (70%) di falsi positivi. Per me, in realtà, sono tutti falsi positivi, anche per tutti gli altri problemi indicati sopra; ma quello che dimostro è che anche dall’interno del paradigma della variante inglese, non si può non ammettere che il 70% dei positivi alla variante sono in realtà falsi positivi.
La falsa pandemia continua, cadendo dalla tragica farsa alla ridicola tragedia, tramite la categoria, ormai divenuta essenziale nella virologia, del “ridicolo scientifico”. Ma virologi, media e governi sono abbastanza tragicamente ridicoli anch’essi per accettare tutto questo senza che li sfiori il minimo dubbio.
L’informazione libera e indipendente ha bisogno del tuo aiuto. Ora più che mai… Database Italia non riceve finanziamenti e si mantiene sulle sue gambe. La continua censura, blocchi delle pubblicità ad intermittenza uniti agli ultimi attacchi informatici non ci permettono di essere completamente autosufficienti.
Fai una donazione seguendo il link sicuro qui sotto
SOSTIENI DATABASE ITALIA